
On December 6, 2023, Novartis reported the approval of Fabhalta® (iptacopan) by the U.S. Food and Drug Administration (FDA) as the first oral monotherapy designed for treating adults with paroxysmal nocturnal hemoglobinuria (PNH). Functioning as a Factor B inhibitor, Fabhalta exerts its effects proximally in the alternative complement pathway of the immune system, ensuring comprehensive control over the destruction of red blood cells (RBC) both within and outside blood vessels (intra- and extravascular hemolysis [IVH and EVH]). Notably, in clinical trials, the administration of Fabhalta resulted in a significant increase in hemoglobin levels (≥ 2 g/dL from baseline in the absence of RBC transfusions) for the majority of patients. Moreover, in the APPLY-PNH study, nearly all patients treated with Fabhalta did not require blood transfusions.
The FDA approval is based on findings from the Phase III APPLY-PNH trial, which involved patients experiencing residual anemia (hemoglobin < 10 g/dL) despite previous anti-C5 treatment. In this trial, individuals who switched to Fabhalta exhibited superior improvement in hemoglobin levels in the absence of RBC transfusions and in transfusion avoidance rate over patients who stayed on anti-C5 treatments. Additionally, Approval was also supported by the Phase III APPOINT-PNH study in complement inhibitor-naïve patients, and the 24-week core treatment periods in the APPLY-PNH and APPOINT-PNH trials, respectively, demonstrated the following outcomes.
- ● 82.3% of patients who had been treated with anti-C5 therapy without transfusion had a sustained increase in hemoglobin levels of ≥2 g/dL after treatment with Fabhalta, and this value was 0% for patients who continued to be treated with anti-C5 therapy (P<0.0001). This result was achieved in 77.5% of patients not treated with complement inhibitors who received Fabhalta.
- ● In the absence of transfusion, 67.7% of patients treated with anti-C5 therapy had sustained hemoglobin levels of ≥12 g/dL, and this value was 0% in patients who continued to be treated with anti-C5 therapy (P<0.0001).
- ● For patients who had been treated with anti-C5 therapy, the rate of avoided transfusions after receiving Fabhalta was 95.2%, while this value was 45.7% for patients in the anti-C5 therapy group (p<0.0001).
About Iptacopan
Iptacopan is a first-in-class oral, small-molecule, reversible inhibitor of factor B, a key serine protease of the alternative pathway of the complement cascade. Iptacopan blocks intravascular hemolysis (IVH) and extravascular hemolysis (EVH) in adults with hemolytic PNH by acting upstream of the C5-terminal pathway of the complement system; it may be able to treat diseases caused by abnormal function of a variety of alternative pathways without affecting the immune response to microbial invasion mediated by other complement pathways, reducing the risk of patients being exposed to infection.

Figure 1. Iptacopan, source: Novartis official website
Iptacopan is presently undergoing clinical development for PNH, as well as for C3 glomerulopathy (C3G) and various other renal conditions characterized by involvement of the complement system, addressing significant unmet needs in conditions such as IgA nephropathy (IgAN), atypical hemolytic uremic syndrome (aHUS), and membranous nephropathy (MN). It holds the potential to emerge as the first inhibitor targeting the complement pathway capable of slowing the progression of diseases driven by the complement system. Based on disease prevalence and the positive interim data from Phase II studies, iptacopan has also received orphan drug designations from both the FDA and EMA for C3G and PNH, along with EMA PRIME designation for C3G and EMA orphan drug designation for IgAN.
About Complement Factor B
The complement system is a vital part of the immune system that plays a key role in defending the body against infections and supporting immune responses. Comprising a complex network of proteins, the complement system acts to recognize, mark, and eliminate pathogens, contributing to immune defense and maintaining overall health.
The complement system can be activated through three distinct pathways:
- ● Classical Pathway: Activated by the binding of antibodies to pathogens, triggering a series of enzymatic reactions.
- ● Alternative Pathway: Constitutively active and continuously surveys foreign invaders, initiating the complement cascade independently of antibodies.
- ● Lectin Pathway: Triggered by the binding of lectins (proteins that recognize carbohydrates) to pathogens, leading to complement activation.
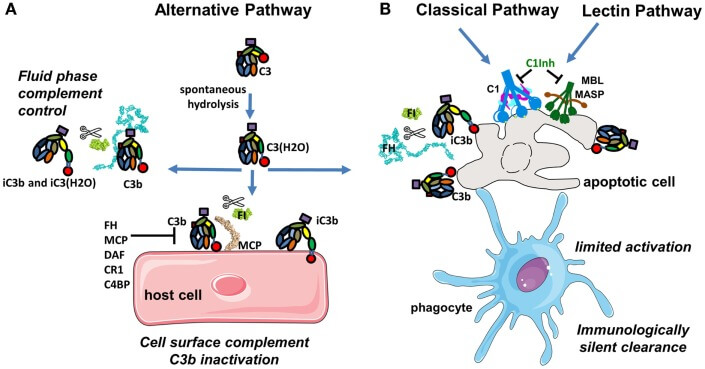
Figure 2. Complement activation pathways [2]
Complement Factor B is a crucial protein in the alternative pathway of the complement system. It combines with other complement proteins to form the C3 convertase enzyme, which plays a central role in cleaving C3 into active fragments. This activation sets off a cascade of events leading to the elimination of pathogens. Factor B's involvement in the alternative pathway contributes to the body's continuous surveillance system, helping to defend against infections and maintain immune balance. Understanding the function of Complement Factor B is essential for developing insights into complement-related diseases and potential therapeutic interventions.
About PNH
PNH is an acquired clonal hematopoietic disorder caused by a mutation in the PIG-A gene on the Xp22.1 locus of somatic cells. Its pathogenesis involves a mutation in the PIG-A gene of hematopoietic stem cells, leading to impaired anchoring of glycosylphosphatidylinositol (GPI) to the cell membrane in part or in full. This results in the loss of GPI-anchored proteins on the surface of blood cells, leading to weakened complement inhibition and increased susceptibility to cell destruction, causing hemolysis, and other related complications. Clinical manifestations primarily include varying degrees of paroxysmal intravascular hemolysis, intermittent hemoglobinuria, bone marrow failure, and venous thrombosis.

Figure 3. Pathogenesis of PNH. [3]
According to statistics, the incidence of PNH is approximately one to two cases per million, with higher rates observed in Asian populations compared to those in Europe and America. PNH can occur at any age but is more common in individuals aged 30-40. Apart from bone marrow transplantation, there is currently no other effective cure for PNH. The primary approach in clinical treatment of this disease is to control hemolytic attacks. Key therapeutic drugs include glucocorticoids, immunosuppressants, complement pathway inhibitors, etc, with anti-complement C5 drugs being the current standard therapy.
Currently, globally approved medications for treating PNH, apart from iptacopan, include only two complement C5 inhibitors: Soliris (eculizumab) and Ultomiris (ravulizumab), both developed by AstraZeneca and administered through intravenous injection.
- ● Soliris: It is the world's first approved complement C5 inhibitor. In addition to its approval for PNH treatment, it is also authorized for treating atypical hemolytic uremic syndrome (aHUS), severe myasthenia gravis (MG) with anti-acetylcholine receptor (AchR) antibody positivity, and neuromyelitis optica spectrum disorder (NMOSD) with aquaporin-4 (AQP4) antibody positive. It is administered weekly or every two weeks. In 2022, its total revenue reached an impressive 3.762 billion USD.
- ● Ultomiris: This is an upgraded version of Soliris. It is currently approved for treating PNH, aHUS (in adults and pediatric patients aged ≥1 month), and generalized myasthenia gravis (gMG). In 2022, its total revenue reached a substantial 1.965 billion USD.
Anti-complement C5 therapy was previously internationally recognized as the standard treatment for PNH, but after anti-complement C5 therapy, a majority of patients still have residual anemia, fatigue, and transfusion dependence, which severely affects quality of life.
References:
[1] https://www.novartis.com/news/media-releases/novartis-receives-fda-approval-fabhalta-iptacopan-offering-superior-hemoglobin-improvement-absence-transfusions-first-oral-monotherapy-adults-pnh
[2] Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol. 2015 Jun 2;6:262. doi: 10.3389/fimmu.2015.00262. PMID: 26082779; PMCID: PMC4451739.
[3] Sahin, Fahri & Comert Ozkan, Melda & Gokmen, Nihal & Yilmaz, Mumtaz & Oruc, Nevin & Gurgun, Alev & Kayikcioglu, Meral & Güler, Ayse & Gokcay, Figen & Bilgir, Ferda & Ceylan, Cengiz & Bilgir, Oktay & Sari, Ismail & Saydam, Guray. (2015). Multidisciplinary clinical management of paroxysmal nocturnal hemoglobinuria. American journal of blood research. 5. 1-9.
Related Articles:
Complement Inhibitors as Therapeutic Agents
Emerging drugs for the treatment of PNH